Genetic Mutation
Category : 12th Class
The idea of mutation first originated from the observations of a Dutch botanist Hugo de Vries (1880) on variations in plants of Oenothera lamarckiana. The mutation can be defined as sudden, stable discontinuous and inheritable variations which appear in organism due to permanent change in their genotype. Mutation is mainly of two types :
(1) Spontaneous mutations : Mutation have been occurring in nature without a known cause is called spontaneous mutation.
(2) Induced mutation : When numerous physical and chemical agents are used to increase the frequency of mutations, they are called induced mutations.
Gene mutations
Gene or point mutations are stable changes in genes i.e. DNA chain. Many times a change in a gene or nucleotide pair does not produce detectable mutation. Thus the point or gene mutation mean the process by which new alleles of a gene are produced. The gene mutation are of following types :
Tautomerism : The changed pairing qualities of the bases (pairing of purine with purine and pyrimidine with pyrimidine) are due to phenomenon called tautomerism.
Tautomeres are the alternate forms of bases and are produced by rearrangements of electrons and protons in the molecules.
Substitutions (Replacements) : These are gene mutations where one or more nitrogenous base pair are changed with others. It may be further of three sub types :
(1) Transition : In transition, a purine (adenine or guanine) or a pyrimidine (cytosine or thymine or uracil) in triplet code of DNA or mRNA is replaced by its type i.e. a purine replaces purine and pyrimidine replaces pyrimidine.
\[GC\to AT\text{ }or\text{ }AT\to GC\]
(2) Transversion : Transversion are substitution gene mutation in which a purine (adenine or guanine) is replaced by pyrimidine (thymine or cytosine) or vice versa.
\[GC\to CG\text{ }or\text{ }TA~~~~,~~~~~AT\to TA\text{ }or\text{ }CG\]
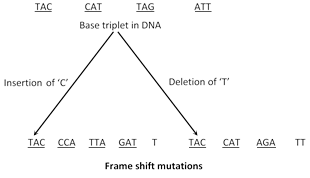
(3) Frame shift mutations : In this type of mutations addition or deletion of single nitrogenous base takes place. None of the codon remains in the same original position and the reading of genetic code is shifted laterally either in the forward or backward direction.
Chromosomal mutation or aberrations
A gene mutation normally alters the information conveyed by a gene, it alters the message. On the other hand, chromosomal mutation only alters the number or position of existing genes. They may involve a modification in the morphology of chromosome or a change in number of chromosomes.
(1) Morphological aberrations of chromosomes
Deletion or deficiency : Sometimes a segment of chromosome break off and get lost. If a terminal segment of a chromosome is lost, it is called deficiency. Deficiency generally proves lethal or semilethal. If intercalary segment is lost it is termed deletion.
![]()
Deletion occurs during pairing in meiosis. For example in human babies deletion of a segment of chromosome number 5 causes a disease called cri-du-chat syndrome (the baby cries like a cat and is mentally retarted with small head).
Wolf-Hirschhorn's syndrome is another well characterized deletion syndrome in human beings caused by a deletion of short arm of chromosome 4 (4p-). The phenotypic effect includes wide-spaced eyes and cleft lip.
Duplication : In this mutation deleted chromosomal segment is attached to its normal homologous chromosome. Here a gene or many genes are repeated twice or more times in the same chromosome.
![]()
Inversion : A piece of chromosome is removed and rejoined in reverse order. For example a chromosome with the gene order A, B, C, D, E, F, G, H is broken between B,C,D and the centre portion turned through 180°, the resulting gene order is A, D, C, B, E, F, G, H.
![]()
Translocation : Mutual exchange (reciprocal) of the chromosome segments between non homologous chromosome. An exchange of parts between two non homologous chromosomes is called reciprocal translocation. In simple translocation a segment of one chromosome breaks and is transferred to another non-homologous chromosome.

(2) Numerical aberrations of chromosomes
Euploidy : The somatic chromosome number in euploids is the exact multiple of basic haploid number. In euploidy an organism acquires an additional set of chromosomes over and above the diploid complement. It can be divided into following types :
(i) Monoploidy or haploidy : Monoploids possess only one set or single basic set of chromosomes. Haploids on the other hand have half the somatic chromosome number. In diploid organisms monoploids and haploids are identical while in a tetra-or hexaploid with 4n or 6n chromosomes the haploids will possess 2n or 3n chromosome whereas its monoploid will possess only one set (n) of chromosome.
(ii) Polyploidy : Organism with more than two sets of chromosomes are known as polyploids. It may be triploid with three sets of chromosomes (3n) or tetraploid with four sets of chromosome (4n) and so on. Polyploidy is of three types:
(a) Autopolyploidy : It is a type of polypoidy in which there is a numerical increase of the same genome, e.g., Autotriploid (AAA), autotetraploid (AAAA). e.g., Maize, Rice, Gram. Autopolyploidy induces gigas effect.
(b) Allopolyploidy : It has developed through hybridisation between two species followed by doubling of chromosomes (e.g., AABB). Allopolyploids function as new species. e.g., Wheat, American cotton, Nicotiana tobacum. Two recently produced allopolyploids are Raphanobrassica and Triticale.
(c) Autoallopolyploidy : It is a type of allopolyploidy in which one genome is in more than diploid state. commonly autoallopolyploids are hexaploids (AAAABB), e.g., Helianthus tuberoseus.
Aneuploidy : Aneuploidy is the term applied for the chromosomal mutations involving only a part of a set, i.e., loss (hypoploidy) or addition (hyperploidy) of one or more chromosomes. Aneuploidy may result from non disjunction of chromosome during cell division.
(i) Monosomy : Diploid organism that are missing one chromosome of a single pair with genomic formula 2n ? 1. Monosomics can form two kind of gametes, (n) and (n ?1). e.g., Turner's syndrome (44+x).
(ii) Nullisomy : An organism that has lost a chromsome pair is nullisomic. The result is usually lethal to diploids \[(2n2).\]
(iii) Trisomy : Diploids which have extra chromosome represented by the chromosomal formula 2n + 1. One of the pairs of chromosomes has an extra member, so that a trivalent may be formed during meiotic prophase. e.g., Down's syndrome (\[45+xx\]or\[45+xy\]), klinefelter's syndrom \[(44+xxy).\] All the possible trisomic have been studied in Datura.
(iv) Tetrasomy : In tetrasomic individual particular chromosome of the haploid set is represented four times in a diploid chromosomal complement. The general chromosomal formula for tetrasomics is \[2n+2\] rather than \[2n+1+1.\] The formula \[2n+1+1\] represents a double trisomic. e.g., Super female \[(44+xxxx).\]
Mutagens
Any substance or agent inducing mutation is called a mutagen. The mutagens may be broadly grouped into two classes:
(1) Physical mutagens : It comprise mainly radiations. Radiation has been used to induce mutations for the first time by H.J. Muller (1927) on animals and L.J. Stadler (1928) on plants. Radiation that can produce mutation is known as effective radiations which are as follows.
(i) Ionizing (Particulate) : a-particles, \[\beta -\]rays, protons and neutrons.
(ii) Ionizing (non particulate) : \[X-\]rays, \[r-\]rays and cosmic rays.
(iii) Nonionizing : Ultraviolet rays
(2) Chemical mutagen : A large number of chemicals react with the four nucleotides and modify their base-pairing capabilities. These are as follows:
(i) Base analogues : 5-bromodeoxyuridine (Brdu), 2-amino purine.
(ii) Chemicals modifying base-pairing
• Hydroxylamine
• Nitrous acid
• Alkylating agent : Nitrogen mustard, ethyl methane sulfonate (EMS), methyl methane sulfonate (MMS) and N-methyl-N-nitro-nitroso-guanidine (NTG).
(iii) Intercalating agents : Proflavin and acridine orange
Genetic diseases in man
There are many diseases in man due to gene mutations. It is either dominant or recessive. The mutated person may become incapable to produce specified enzyme, so result in inborn errors of metabolism.
Chondrodystrophic dwarfism : Chondrodystrophic dwarfism is a dominant autosomal mutation, most people are homozygous for recessive allele (c/c). The presence of one dominant C results in the premature closure of the growth areas of long bones of arms and legs, resulting in shortened and bowed arms and legs.
Huntington disease : Huntington disease is caused by a dominant gene on chromosome 4. The mutated gene causes abnormality by producing a substance that interferes with normal metabolism in the brain that leads to progressive degeneration of brain cells. The death comes ten to fifteen years after the onset of symptoms.
Neurofibromatosis : Also called ?von Recklinghausen disease? caused by a dominant gene on chromosome 17. The affected individual may have ten spots on the skin which later may increase in size and number. Small benign tumours called neurofibromas may occur under the skin or in various organs.
Tay-Sachs disease : Tay-Sachs disease results from the lack of the dominant gene on chromosome 15 for the production of hexosaminidase and subsequent storage of its substrate, a fatty substance known as glycosphingolipid, in lysosomes. The patient suffers from defective vision, muscular weakness and gradual loss of all mental and physical control, death occurs by the age of three or four years.
Cystic fibrosis : The most common lethal genetic disease due to a recessive mutation on the chromosome 7. The body produces abnormal glycoprotein which interferes with salt metabolism. he mucus secreted by body becomes abnormally viscid and blocks passages in the lungs, liver and pancreas.
Alzheimer's disease : Alzheimer's disease, named after the German neurologist Alzheimer, is a degenerative brain disease characterized by memory loss, confusion, restlessness, speech disturbances, erosion of personality, judgement, and inability to perform the functions of daily living. Alzheimer's disease, a form of dementia, occurs in karyotypically normal individuals. The brain of Alzheimer's patients show a marked loss of neurons. These patients also show an accumulation of senile plaques, which are thickened nerve cell processes (axons and dendrites) surrounding a deposit of particular type of polypeptide called amyloid \[\beta \] protein. The occurrence of Alzheimer's disease in people with Down's syndrome suggests that a gene or genes on chromosome 21 is involved. According to Bush (2003) Alzheimer's disease is caused by a copper and zinc build up in the brain.
Marfan's syndrome : Marfan's syndrome is due to dominant mutation resulting in the production of abnormal form of connective tissues and characteristic extreme looseness of joints. The long bones of body grow longer, fingers are very long called 'spider fingers' or arachnodactyly. The lenses in eyes become displaced.
Albinism : Albinism is an autosomal recessive mutation. An albino cannot synthesize melanin which provides black colouration to skin and hair. Albinism is due to tyrosinase deficiency. The enzyme tyrosinase normally converts the amino acid tyrosine to melanin through an intermediate product DOPA (dihydro phenyl alanine).
Sickle-cell disease : Sickle-cell disease is a genetic disease reported from negroes due to a molecular mutation of gene \[H{{b}^{A}}\]on chromosome 11 which produces the \[\beta \] chain of adult haemoglobin. The mutated gene \[H{{b}^{S}}\] produces sickle-cell haemoglobin. The sixth amino acid in \[\beta \] chain of normal haemoglobin is glutamic acid. In sickle-cell haemoglobin this amino acid is replaced by valine. The children homozygous \[(H{{b}^{S}}H{{b}^{S}})\] produce rigid chains. When oxygen level of the blood drops below certain level, RBCs undergo sickling. Such cells do not transport oxygen efficiently; they are removed by spleen causing severe anaemia. Individuals with the \[H{{b}^{A}}H{{b}^{A}}\] genotype are normal, those with the \[H{{b}^{S}}H{{b}^{S}}\]genotype have sickle-cell disease, and those with the \[H{{b}^{A}}H{{b}^{S}}\]genotypes have the sickle-cell trait. Two individuals with sickle-cell trait can produce children with all three phenotypes. Individuals of sickle-cell trait are immune to malaria.
Thalassemia : Thalassemia is a human anaemia due to an autosomal mutant gene and when this gene is present in double dose, the disease is severe thalassemia major with death occurring in childhood. Heterozygous persons show a milder disease, thalassemia minor or also called Cooley's anaemia. The persons suffering from thalassemia major are unable to produce \[\beta \] chain. Their haemoglobin contains \[\delta \] chains like that of foetus which is unable to carry out normal oxygen transporting function.
Alkaptonuria : Alkaptonuria was the first of the recessive human trait discovered in 1902 by Archibald Garrod, ?father of physiological genetics 'or' father of biochemical genetics'. Patients of alkaptonuria excrete large amounts of homogentistic acid in urine. Such urine turns black upon exposure to light. In normal person, homogentistic acid (alkapton) is oxidized by a liver enzyme homogentistic acid oxidase to maleyl acetoacetic acid.
Phenylketonuria (PKU) : Phenylketonuria was discovered by the Norwegian physician A. Folling in 1934; an autosomal recessive mutation of gene on chromosome 12. PKU results when there is a deficiency of liver enzyme phenylalanine hydroxylase that converts phenylalanine into tyrosine. There is a high level phenylalanine in their blood and tissue fluids. Increased phenylalanine in the blood interferes with brain development; muscles and cartilages of the legs may be defective and the patients cannot walk properly.
Gaucher's disease : Gaucher's disease is a genetic disease associated with abnormal fat metabolism, caused by the absence of the enzyme glucocerebrosidase required for proper processing of lipids. Non processing of lipids results in accumulation of fatty material in spleen, liver, bone marrow and brain. The swelling of these organs occurs and patients usually die by the age of 15 years.
Galactosemia : Galactosemia is inherited as an autosomal recessive, and the affected person is unable to convert galactose to glucose. Galactosemia is due to the deficiency of the enzyme Galactose Phosphate uridyl Transferase (GPT). Milk is toxic to galactosemic infants; child usually dies at three years of age.
Taste blindness of PTC : Taste blindness of PTC is a genetic trait, not a disease, discovered by Fox in 1932. PTC (phenyl thiocarbamide) is a compound of nitrogen, carbon and sulphur with sour taste. About 30% people lack the ability to taste PTC which is transmitted by a dominant gene T. The genotypes TT and Tt are tasters of PTC, while tt are non-tasters or taste blind persons.
Chronic Myelogenous Leukaemia (CML) : Chronic myelogenous leukaemia in human beings is a fatal cancer involving uncontrolled replication of myeloblasts (stem cells of white blood cells). Ninety percent of CML is associated with an aberration of chromosome 22. This abnormal chromosome was originally discovered in the city of Philadelphia in 1959 and thus is called the 'Philadelphia chromosome'. In the Philadelphia translocation, the tip of the long arm of chromosome 9 has been joined to the body of chromosome 22 and the distal portion of the long arm of chromosome 22 has been joined to the body of chromosome 9. CML is characterized by an excess of granular leucocytes in the blood. With the increase in the number of leucocytes, there is a reduction in the number of RBCs resulting in severe anaemia.
Burkitt's Lymphoma : Burkitt's lymphoma, a particularly common disease in Africa, is another example of a white blood cell cancer associated with reciprocal translocations. These translocations invariably involve chromosome 8 and one of the three chromosomes (2, 14 and 22) that carry genes encoding the polypeptides that form immunoglobulins or antibodies. Translocations involving chromosomes 8 and 14 are the most common.
Sex chromosome abnormalities
Turner's syndrome : Such persons are monosomic for sex chromosomes i.e. possess only one X and no Y chromosome (XO). In other words they have chromosome number \[2n1=45.\] They are phenotypic females but are sterile because they have under developed reproductive organs. They are dwarf about 4 feet 10 inches and are flat chested with wide spread nipples of mammary glands which never enlarge like those in normal woman. They develop as normal female in childhood but at adolescence their ovaries remain under developed. They lack female hormone estrogen. About one out of every 5,000 female births results in Turner's syndrome.
Klinefelter's syndrome : Since 1942, this abnormality of sex is known to geneticists and physicians. It occurs due to Trisomy of sex chromosomes which results in (XXY) sex chromosomes. Total chromosomes in such persons are \[2n+1=47\] in place of 46. Klinefelter (1942) found that testes in such male remain under developed in adulthood. They develop secondary sex characters of female like large breasts and loss of facial hair. Characters of male develop due to Y chromosome and those like female due to XX chromosomes. About one male child out of every 5,000 born, develops Klinefelter's syndrome.
Such children are born as a result of fertilization of abnormal eggs (XX) by normal sperms with (X) or (Y) chromosomes or by fertilization of normal eggs with (X) chromosomes by abnormal sperms with (XY) chromosome. They are sterile males mentally retarded and are eunuchs.
Super females or metasuper females : Presence of extra (X) chromosomes in females shows such condition leading to (XXX, XXXX, XXXXX), having total 47, 48 or 49 chromosomes in each cell. Females with this type of aneuploidy show abnormal sexual development and mental retardation. Severeness of abnormality increases with the increase in number of (X) chromosomes.
Criminal's or Jacob's syndrome (super males) : Presence of an extra (Y) chromosome in males causes such a condition (XYY) resulting in individuals with \[2n+1=47\] chromosomes. They have unusual height, mentally retarded and criminal bent of mind since birth. Their genital organs are under developed. Their frequency is one in every 300 males.
Autosomal abnormalities
Down's syndrome : This autosomal abnormality is also known as Mongolian idiocy or mongolism. In Langdon Down of England (1866) studied the Mongolian idiocy and described the trisomic condition of their chromosomes. Down's syndrome, a very common congenital abnormality arises due to the failure of separation of 21st pair of autosomes during meiosis. Thus an egg is produced with 24 chromosomes instead of 23. A Down's syndrome has 3 autosomes in 21st pair instead of 2. Total number of chromosomes in this case is \[2n+1\,({{21}^{st}})=47.\]
The affected children have a very broad fore head, short neck, flat palms without crease, stubby fingers, permanently open mouth, projecting lower jaw and a long thick extending tongue. They have low intelligence and are short heighted. They have defective heart and other organs. They are born to mothers aged 40 year and above during first pregnancy. They may survive upto 20 years under medical care.
They are called mongolian idiots because of their round, dull face and upper eyelids stretched downwards similar to mongolian race.
Edward's syndrome : This autosomal abnormality occurs due to trisomy of eighteenth pair of autosomes in which the number of chromosomes are \[2n+1=47.\]The child with this defect survives only about 6 months. Such children have defective nervous system, malformed ears and a receding chin.
Patau's syndrome : This is trisomy of thirteenth pair of autosomal chromosome. This trisomic condition involves numerous malformations such as harelip, clefted palate and cerebral, ocular and cardiovascular defects. Such children usually survive for about 3 months only.




You need to login to perform this action.
You will be redirected in
3 sec

